Goa is abuzz with excitement as vintage bike and car owners, users, collectors and fans are decking […]
Old is Gold
October 4, 2016

August may be known for its monsoon rains and vibrant festivals, but for the medical community, it carries a deeper significance – it is the Spinal Muscular Atrophy (SMA) Awareness Month. This rare genetic condition quietly weakens muscles, yet reveals the extraordinary strength of those who live with it. “Awareness is the first step to action,” says Dr Amit Dias, as he connects the dots for us and explains what can be done to support patients and families….
IMAGINE watching your child smile brightly but struggle to lift his or her head, sit, or take the first steps of life. For families affected by Spinal Muscular Atrophy (SMA), this is a painful reality. SMA is a rare yet devastating genetic disorder that robs children of the ability to move, eat or even breathe as motor neurons in the spinal cord gradually weaken and die. This August, observed globally as SMA Awareness Month, carries the theme “Connecting the Dots” — reminding us that awareness, early detection, access to treatment, and community support are all vital pieces of the puzzle in giving these children a fighting chance.
What Causes SMA? – The Genetic Story
SMA is caused by a defect in a gene called SMN1 (Survival Motor Neuron 1). This gene produces a protein essential for the survival of motor neurons. Without enough of this protein, motor neurons degenerate.
Inheritance: SMA is an autosomal recessive disorder – a child must inherit two faulty copies of the SMN1 gene (one from each parent) to develop the condition.
Carrier status: Parents carrying a single faulty gene do not have symptoms, but there’s a 25% chance with each pregnancy that their child will have SMA if both parents are carriers.
How is SMA Detected?
Newborn screening: In many countries, SMA is now detected soon after birth through a simple blood test as part of newborn screening programs.
Genetic testing: If a child shows early symptoms, a genetic test can confirm the diagnosis by identifying changes in the SMN1 gene.
Prenatal diagnosis: For high-risk families, prenatal genetic testing is possible during pregnancy.
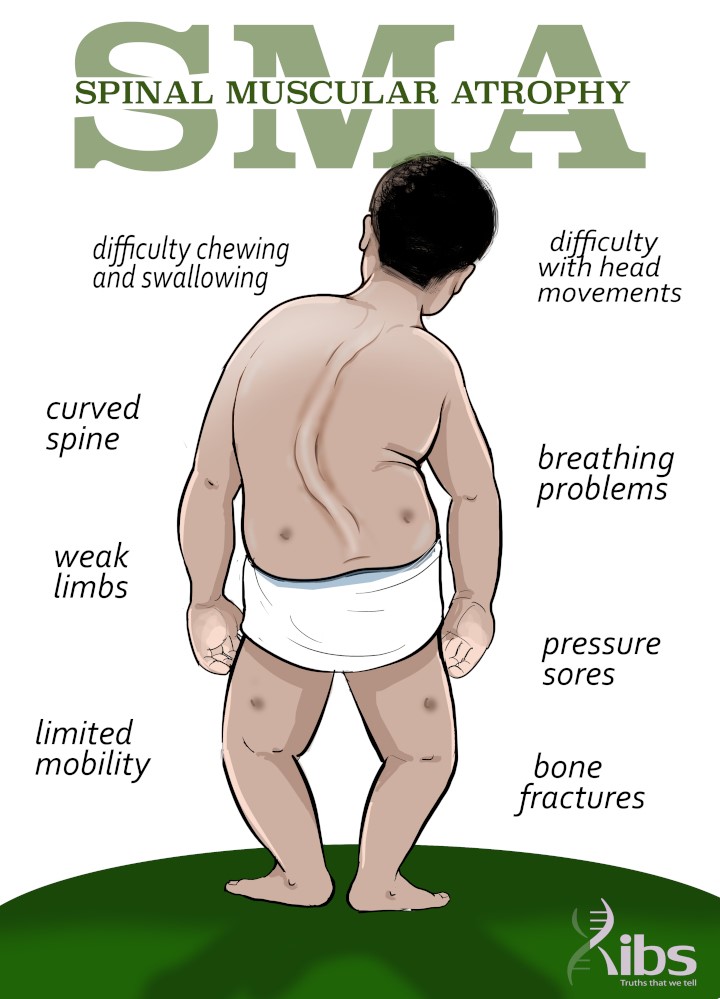
Signs and Symptoms
The severity and age of onset vary, but symptoms often include:
• Muscle weakness (often starting in the legs and progressing upwards)
• Difficulty holding the head up in infants
• Delayed motor milestones (rolling, sitting, walking)
• Weak cry or cough
• Breathing difficulties
• Swallowing problems
Types and Progression
Types of Spinal Muscular Atrophy (SMA)
Spinal Muscular Atrophy is classified into types based on the age of onset and the severity of motor impairment:
Type 0 (Prenatal SMA): The rarest and most severe form, identified during pregnancy by reduced fetal movements. At birth, infants present with profound muscle weakness, hypotonic, joint contractures, and minimal response to external stimuli. Respiratory failure is common, and survival is usually less than six months.
Type 1 (Werdnig–Hoffmann disease): The most common form, with onset between birth and 6 months of age. Affected infants never achieve the ability to sit independently. They exhibit severe hypotonia (a condition characterized by decreased muscle tone), weak cry, feeding difficulties, and respiratory distress. Without treatment, survival rarely extends beyond 2 years, primarily due to respiratory complications.
Type 2 (Intermediate SMA or Dubowitz disease): Symptoms appear between 6 and 18 months. Children can usually sit independently but cannot stand or walk unaided. Progressive muscle weakness and scoliosis are common. With supportive care, survival often extends into the second or third decade of life, though respiratory complications may shorten lifespan.
Type 3 (Kugelberg–Welander disease): Onset occurs after 18 months of age, sometimes in late childhood or adolescence. Affected individuals achieve independent walking, though they may lose this ability over time due to progressive weakness. Respiratory involvement is less severe, and life expectancy is typically normal.
Type 4 (Adult-onset SMA): Symptoms begin in early adulthood (usually in the second or third decade). This form is the mildest, characterized by gradual, proximal muscle weakness—especially in the legs. Most individuals retain mobility and respiratory function, and life expectancy is unaffected.
Without treatment, SMA often leads to progressive disability, respiratory problems, and dependence on assistive devices.
What Can Be Done? – The Hope in Treatment
The last decade has brought revolutionary changes in SMA management.
Disease-modifying therapies:
Nusinersen – injected into the spinal fluid, helps the body produce more SMN protein.
Onasemnogene abeparvovec – a gene therapy delivering a healthy SMN1 gene.
Risdiplam – an oral medicine that increases SMN protein production.
These treatments work best when started early, even before symptoms appear.
Supportive care:
Respiratory support (ventilation, cough assist devices)
Nutritional support and swallowing therapy
Physiotherapy and occupational therapy to maintain movement and prevent contractures
Assistive devices like wheelchairs, braces, and adaptive seating.
What Do Patients and Families Need?
Early diagnosis: Screening programs are crucial.
Access to treatment: High costs remain a major challenge.
Multidisciplinary care: Neurologists, physiotherapists, pulmonologists, nutritionists, and psychologists all play a role.
Emotional support: SMA affects the whole family – counseling and peer support are invaluable.
How Big is the Problem?
SMA affects approximately 1 in 6,000–10,000 births worldwide.
It is a leading genetic cause of death in infants.
In India, thousands of children live with SMA, many without access to the latest treatments due to cost barriers.
The Way Forward
With advances in genetic medicine, SMA no longer has to be a death sentence. Early detection, timely treatment, and comprehensive care can help children live longer, healthier, and more independent lives. As a society, we must work together to:
Raise awareness about SMA and genetic disorders
Advocate for newborn screening programs
Support families emotionally and financially
Push for affordable access to life-saving therapies
In conclusion:
Spinal Muscular Atrophy is a challenging condition, but science is giving us tools to fight it. Our mission should be to ensure that no child is left behind due to lack of awareness or resources. Every child with SMA deserves the chance to move, to play, and to live life to the fullest.

 LETTER TO THE EDITOR FOR ISSUE DATED APRIL 11, 2026
LETTER TO THE EDITOR FOR ISSUE DATED APRIL 11, 2026 DOUBLE ENGINE GOA GOVERNMENT DANCES TO ADANI TUNES!
DOUBLE ENGINE GOA GOVERNMENT DANCES TO ADANI TUNES! WHO IS FIXING THE SKY? The billionaires, the cops in SUVs, and the missing trees! By Raaisa Lemos Vaz
WHO IS FIXING THE SKY? The billionaires, the cops in SUVs, and the missing trees! By Raaisa Lemos Vaz UPHEAVAL OF THE SUSEGAAD!By Sumaiya Khan
UPHEAVAL OF THE SUSEGAAD!By Sumaiya Khan THE POWER AND THE POLITICS OF MINING IN GOA!By Arvind Pinto
THE POWER AND THE POLITICS OF MINING IN GOA!By Arvind Pinto DEVA VANI DHRUPAD: Alchemy of Sound, Silence and Sanctity!By Prema Viswanathan
DEVA VANI DHRUPAD: Alchemy of Sound, Silence and Sanctity!By Prema Viswanathan AT LEAST HOSPITALS CAN OFFER MILLET ROTI AND KICHADI!
AT LEAST HOSPITALS CAN OFFER MILLET ROTI AND KICHADI! WHEN DO WE NEED PALLIATIVE CARE?
WHEN DO WE NEED PALLIATIVE CARE? GODI MEDIA TRUMPS TRUTH IN WAR & PEACE!
GODI MEDIA TRUMPS TRUTH IN WAR & PEACE! World Health Day 2026: TOGETHER FOR HEALTH – STAND WITH SCIENCE! By Dr Amit Dias, MD
World Health Day 2026: TOGETHER FOR HEALTH – STAND WITH SCIENCE! By Dr Amit Dias, MD FCRA AMENDMENT WILL ENABLE GOVT TO TAKE OVER PROPERTIES!By Olav Albuquerque
FCRA AMENDMENT WILL ENABLE GOVT TO TAKE OVER PROPERTIES!By Olav Albuquerque WEEKEND UPDATESLETTERS TO THE EDITOR FOR ISSUE DATED APRIL 04, 2026
WEEKEND UPDATESLETTERS TO THE EDITOR FOR ISSUE DATED APRIL 04, 2026 REAL ESTATE BARONS OF GOA!
REAL ESTATE BARONS OF GOA! THE SLOW VANISHING OF GOA’S GOLDEN BEACHES! By Raaisa Lemos Vaz
THE SLOW VANISHING OF GOA’S GOLDEN BEACHES! By Raaisa Lemos Vaz
Goa is abuzz with excitement as vintage bike and car owners, users, collectors and fans are decking […]